
INFORMAȚII
Ce este hemofilia
Hemofilia (din limba greacă haima αἷμα ‘sânge’ și philia φιλία ‘dragoste’) este o boală ereditară, transmisă de obicei de mamă. Este mult mai frecventă la băieți decât la fete și constă într-o predispoziție spre hemoragie, cauzată de prelungirea timpului de coagulare a sângelui. Ea este un sindrom hemoragipar sever care apare la copiii de sex masculin și este dependent de transmiterea unei gene anormale legată de cromozomul „X”. Hemofilia A (deficiența coagulării factorului VIII) este cea mai răspândită formă a bolii, prezentă la aproximativ 1 din 5.000-10.000 de copii născuți de sex masculin. Hemofilia B (deficiența factorului IX) apare la aproximativ 1 din 20.000-34.000 de nou-născuți de sex masculin.
 Termenul de hemofilie a fost introdus prima dată de Schönlein în 1893, deși boala era cunoscută încă din antichitate, fiind descrisă de Rabbi Simon ben Gamaliel în Talmud, de Maimonide (medic și filozof evreu), de Albucasis, medic arab (în secolul al XII-lea), Addis (1911), Patek și Taylor în 1937 – ei au demonstrat prezența unui factor activ antihemofilic în plasma normală.
Termenul de hemofilie a fost introdus prima dată de Schönlein în 1893, deși boala era cunoscută încă din antichitate, fiind descrisă de Rabbi Simon ben Gamaliel în Talmud, de Maimonide (medic și filozof evreu), de Albucasis, medic arab (în secolul al XII-lea), Addis (1911), Patek și Taylor în 1937 – ei au demonstrat prezența unui factor activ antihemofilic în plasma normală.
În aproximativ jumătate din cazuri – hemofilia este o boală ereditară – se transmite din generație în generație prin linia feminină. Iar a doua jumătate este rezultatul mutațiilor genetice aleatorii. Secretul lor este încă necunoscut nimănui din lume. De aceea, în viitorul apropiat nu vom putea face parte cu hemofilie. Cea mai mare problemă este că acestea sunt mutații aleatorii, spontane. Este imposibil să le prezicem. Prin urmare, este imposibil de avertizat.
Hemofilia-simptome, diagnostic, tratament

Hemofilia este o boală genetică ce afectează capacitatea sângelui de a se coagula. Cu alte cuvinte, hemofilia reprezintă o coagulopatie. În mod caracteristic, hemofilia presupune deficitul factorilor coagulării VIII sau IX. La persoanele cu hemofilie perioada de sângerare se prelungește, în funcție de cât de sever este deficitul de factor de coagulare.
Hemofilia afectează persoanele de sex masculin și doar în mod excepțional persoanele de sex feminin, la acestea formele de boală fiind mult mai blânde.
Cauzele hemofiliei
Gena defectă, responsabilă pentru deficitul de factor de coagulare VIII sau IX, se găsește pe cromozomul X. După cum știm, persoanele de sex feminin dețin în codul lor genetic doi cromozomi X. Cele de sex masculin, dețin un cromozom X și un cromozom Y. Dacă o fată are un cromozom X purtător al genei responsabile de hemofilie, are și un cromozom X normal, care va fi capabil să compenseze deficiența perechii sale. Băieții, in schimb, având un singur cromozom X, nu au cu ce compensa anormalitatea acestuia: cromozomul Y nu are proprietăți similare.
Tipuri de hemofilie
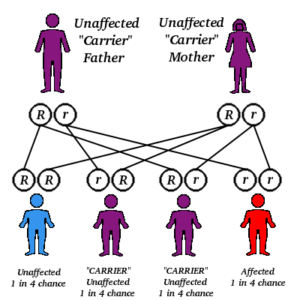 Hemofilia A, sau hemofilia clasică, reprezintă deficiența de factor VIII. Acesta este cel mai întâlnit tip de hemofilie, fiind de 5 ori mai frecventă decât hemophilia B. Incidența este de 1:10 000 nou-născuți.
Hemofilia A, sau hemofilia clasică, reprezintă deficiența de factor VIII. Acesta este cel mai întâlnit tip de hemofilie, fiind de 5 ori mai frecventă decât hemophilia B. Incidența este de 1:10 000 nou-născuți.
Hemofilia B, sau Boala Christmas, reprezintă deficiența de factor IX. Incidența este de 1:50 000 nou-născuți.
Simptomele hemofiliei
Severitatea manifestărilor clinice depinde de gradul deficitului de factor de coagulare. Astfel, putem avea de a face cu forme ușoare, medii sau severe. Dacă deficitul este sever, pacientul poate avea sângerări sponane.
 Simptomul caracteristic, cel care ridică suspiciunea de hemofilie, este sângerarea prelungită. Aceasta poate fi externă, vizibilă ca: epistaxis, sângerare prelungită în urma unei tăieturi sau zgârieturi, sângerare prelungită în urma unor extracții dentare. De cele mai multe ori, însă, sângerarea este internă, obiectivându-se ca hematurie, melenă, hematochezie, echimoze extinse.
Simptomul caracteristic, cel care ridică suspiciunea de hemofilie, este sângerarea prelungită. Aceasta poate fi externă, vizibilă ca: epistaxis, sângerare prelungită în urma unei tăieturi sau zgârieturi, sângerare prelungită în urma unor extracții dentare. De cele mai multe ori, însă, sângerarea este internă, obiectivându-se ca hematurie, melenă, hematochezie, echimoze extinse.
Frecvent, sediul sângerării spontane sau după traumatisme minime este reprezentat de articulații, rezultând hemartroză. În timp, hemartrozele repetate, datorită efectului iritant al sângelui asupra sinovialei, pot determina inflamația și proliferarea acesteia, proces care evoluează către artropatie hemofilică.
Diagnosticul hemofiliei
 De foarte multe ori manifestările hemofiliei apar din copilărie, odată ce copilul începe să se deplaseze de unul singur. În acest caz, se pot observa echimozele și sângerările la traumatisme minore. Aceste manifestări ridică suspiciunea de hemofilie. Dacă în familie există perosane cu hemofilie confirmată sau cu manifestări asemănătoare, diagnosticul este cu atât mai probabil. Cu alte cuvinte, istoricul pacientului și antecedentele heredo-colaterale joacă un rol esențial, episoadele de sângerări prelungite sau colecții hematice intraarticulare orientând diagnosticul.
De foarte multe ori manifestările hemofiliei apar din copilărie, odată ce copilul începe să se deplaseze de unul singur. În acest caz, se pot observa echimozele și sângerările la traumatisme minore. Aceste manifestări ridică suspiciunea de hemofilie. Dacă în familie există perosane cu hemofilie confirmată sau cu manifestări asemănătoare, diagnosticul este cu atât mai probabil. Cu alte cuvinte, istoricul pacientului și antecedentele heredo-colaterale joacă un rol esențial, episoadele de sângerări prelungite sau colecții hematice intraarticulare orientând diagnosticul.
Fiind o coagulopatie, apar modificări ale coagulogramei. Caracteristic hemofiliei este creșterea timpului de trombolpastină parțială activată (aPTT). Numărul de trombocite și timpul de protrombină au valori în limite normale.
Dozarea factorilor de coagulare în sânge este esențială deoarece stabilește factorul care se află în deficit, deci determină dacă hemofilia este de tip A (factorul VIII) sau B (factorul IX), informație crucială pentru instituirea tratamentului de substituție. De asemenea, apreciază cantitativ deficitul, deci apreciază severitatea bolii.
Tratamentul hemofiliei
 Principala modalitate de tratament la momentul actual este reprezentată de terapia de substituție. Cu alte cuvinte deficitul endogen de factori de coagulare este compensate prin aport exogen, astfel încât se administrează o soluție concentrată de factor VIII, pentru hemofilia A, sau factor IX, pentru hemofilia B. Aceste preparate de factor VIII sau IX sunt injectabile și pot fi de origine umană sau sintetică.
Principala modalitate de tratament la momentul actual este reprezentată de terapia de substituție. Cu alte cuvinte deficitul endogen de factori de coagulare este compensate prin aport exogen, astfel încât se administrează o soluție concentrată de factor VIII, pentru hemofilia A, sau factor IX, pentru hemofilia B. Aceste preparate de factor VIII sau IX sunt injectabile și pot fi de origine umană sau sintetică.

Tratamentul de substituție se poate administra atât în cazul unui episod acut de sângerare, pentru a opri sângerarea, dar și în scop profilactic, în spital sau la domiciliu. Schema și dozele de administrare depind de severitatea bolii, în formele severe fiind necesară administrarea pe toată durata vieții pacientului.
În cazul sângerării intraarticulare se impune evacuarea sângelui pentru a evita dezvoltarea artropatiei hemofilice. Puncția evacuatorie poate fi însoțită de tratament simptomatic, cum ar fi medicamente din clasa analgezicelor, antiinflamatoarelor sau corticosteroizilor.
 Cum se manifestă hemofilia.
Cum se manifestă hemofilia.
La copiii mici, hematoamele apar adesea pe cap, în zona feselor, cu lopeți. Dintii fiziologici sunt insotiti de sangerare continua. Adesea, există, de asemenea, o descărcare de sânge din mucoasele nasului și gurii atunci când striviți limba și obrajii.
Leziunile ochilor sunt deosebit de periculoase. Sângerarea puternica poate duce la orbire completă.
Odată cu vârsta, manifestările devin moderate, sângerarea este netezită, pericolul lor nu mai este atât de mare.
 În viața de zi cu zi, există un mit al cazurilor de sângerare la pacienții cu hemofilie care suferă de cea mai mică excizie sau zgârietură. De fapt, nu este. Chirurgia gravă și sângerarea internă de origine neexplicată sunt periculoase. Cel mai probabil, o combinație de mecanisme de sângerare cu boală și fragilitate, permeabilitatea peretelui vascular.
În viața de zi cu zi, există un mit al cazurilor de sângerare la pacienții cu hemofilie care suferă de cea mai mică excizie sau zgârietură. De fapt, nu este. Chirurgia gravă și sângerarea internă de origine neexplicată sunt periculoase. Cel mai probabil, o combinație de mecanisme de sângerare cu boală și fragilitate, permeabilitatea peretelui vascular.
 Notă: Sângerare recurentă după accidentare a fost observată la pacienții cu hemofilie. Pe fundalul unei opriri, în câteva ore sau zile, procesul poate fi repetat.Prin urmare, este necesar să monitorizați cu atenție astfel de pacienți. Sângerarea recurentă frecventă provoacă anemie în timp.
Notă: Sângerare recurentă după accidentare a fost observată la pacienții cu hemofilie. Pe fundalul unei opriri, în câteva ore sau zile, procesul poate fi repetat.Prin urmare, este necesar să monitorizați cu atenție astfel de pacienți. Sângerarea recurentă frecventă provoacă anemie în timp.
 |
Motto: “Hemofilia nu așteaptă să se facă dimineaţă !” |
Tags
Haemophilia, doctor, blood, coagulation-diagnosis, cromosome-mutation, science-blood, donation, healt-medicine, splashing-person-location, plabelebs, bleed, slimy-bleed-clinical-white, clinic-profesional-suport, disorder, dragstore-medic-condition, red-illnes, job, flow, physician, hemorhage, rhesus-aneamia-splattered-knowledge-help, apothecary, pharmacy-medical, hepatitis, donor-practice, clot, assistant-dripping, platelebs-flowing, doneaza, hemoragie, redirectionare2%, hemofilie fundatie, asociatia hemofilia.